
Pompe Disease
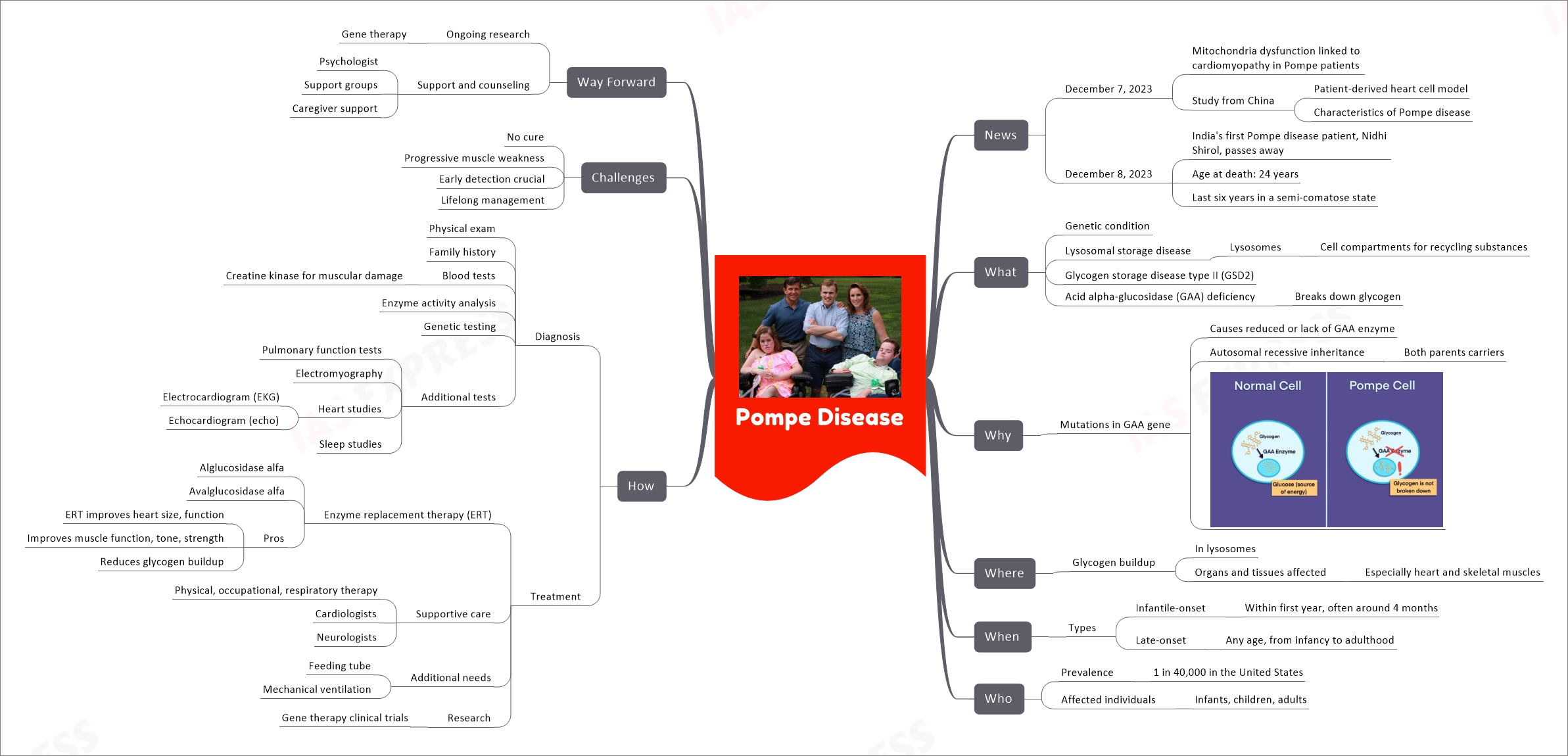
| Category | Subcategory | Details |
|---|---|---|
| News | December 7, 2023 | Mitochondria dysfunction linked to cardiomyopathy in Pompe patients |
| Study from China | ||
| Patient-derived heart cell model | ||
| Characteristics of Pompe disease | ||
| December 8, 2023 | India’s first Pompe disease patient, Nidhi Shirol, passes away | |
| Age at death: 24 years | ||
| Last six years in a semi-comatose state | ||
| What | Genetic condition | Lysosomal storage disease |
| Lysosomes as cell compartments | ||
| Glycogen storage disease type II (GSD2) | ||
| Acid alpha-glucosidase (GAA) deficiency | ||
| Why | Mutations in GAA gene | Causes reduced or lack of GAA enzyme |
| Autosomal recessive inheritance | ||
| Both parents carriers | ||
| Where | Glycogen buildup | In lysosomes |
| Organs and tissues affected, especially heart and skeletal muscles | ||
| When | Types | Infantile-onset: Within first year, often around 4 months |
| Late-onset: Any age, from infancy to adulthood | ||
| Who | Prevalence | 1 in 40,000 in the United States |
| Affected individuals | Infants, children, adults | |
| How | Diagnosis | Physical exam, family history |
| Blood tests for muscular damage | ||
| Enzyme activity analysis | ||
| Genetic testing | ||
| Additional tests: Pulmonary function tests, Electromyography, Heart studies (EKG, echo), Sleep studies | ||
| Treatment | Enzyme replacement therapy (ERT): Alglucosidase alfa, Avalglucosidase alfa | |
| Supportive care: Physical, occupational, respiratory therapy; Cardiologists; Neurologists | ||
| Additional needs: Feeding tube, Mechanical ventilation | ||
| Research: Gene therapy clinical trials ERT improves heart size, function, muscle function, tone, strength, reduces glycogen buildup | ||
| Challenges | Early detection crucial | Lifelong management |
| No cure | Progressive muscle weakness | |
| Way Forward | Ongoing research | Gene therapy |
| Support and counseling: Psychologist, Support groups, Caregiver support |
Related Posts
If you like this post, please share your feedback in the comments section below so that we will upload more posts like this.

Responses